解析結果
|
CrystalClearの結果は、「E:\01CrystalClear_Data」に保存されている。
|
 (図2.1) CrystalClear実行ファイル |
これらの多くのファイルの中で、以下の2つが直接的に重要。
-
“dtscaleaverage.log” … “Scale and Average” の実行結果。公表時に一般的に必要とされる情報がまとめられている。
- “ScalAveraged.ref” … 立体構造解析に用いる反射ファイル(“Scale and Average” 実行時に任意の名称を与えていれば、それ)。
参考:dtscaleaverage.log
“dtscaleaverage.log”最後の“Summary of data collection statistics”が回折実験のまとめ(図2.2)。うち下のパラメータは公表時にも必要となる(どれが必要となるかは、Journal次第)。
|
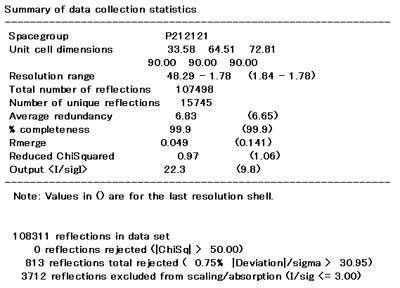 (図2.2) “Scale and Average”結果 |
各値の良し悪しの判断基準は、“
CrystalClear ver.1.3 SP1 / 解析終了後 / その他
”を参照のこと。
立体構造解析へ
複数の反射データの作製
状況によっては、複数の反射データを作成する(=“Scale and Averaged”を繰り返す)必要があるため、以下、記述しておく。
なお“Scale and Averaged”を繰り返し実行する際は、“ScalAveraged.ref”を任意の名称に変更して実行すること。
加えて「E:\01CrystalClear_Data」内の“dtscaleaverage.log”を“名称の変更”しておく(同じファイル名だと上書きされる)。
分解能を変更
default設定では、回折画像撮像時のカメラ長に依存した分解能で反射データが作製されるが、
最外郭のデータが悪い場合(R-mergeが0.2以上)など、その部分をカットした反射データを併せて作成しておく。
“Scale and Average” 実施時に、“Resolution”の“Maxinum”を変更すれば良い。
MIR法・SAD法
MIR法・SAD法では、anomalousの情報入りの反射データを作成し、それを位相決定に用いる必要がある。
ただし、位相決定後の構造精密化の過程では、anomalous情報を抜いた反射データを用いるのが一般的であるため、
それに対応した反射データを別途作成しておく。
簡単に言えば、“Scale and Average”を
・“Scale I+ and I- separately”、“Outout anomalous (I+,I-)”: それぞれチェック有 … 位相決定に利用
・“Scale I+ and I- separately”、“Outout anomalous (I+,I-)”: それぞれチェック無 … 構造精密化に利用
とパラメータ設定を変更して2回実行し、それぞれrefファイルを作成する。
空間群が未決定
“Analyze Data”・“Space Group Check”で空間群が決定できない場合が稀にある
(参考:“Space Group Results”)。
その場合、可能性のある空間群それぞれで“Scale and Average” 実施し、反射データを作成しておく。
実際に正しい空間群がどれかは、それぞれの反射データで構造解析を実施して確認する(簡単に言えば、位相が決定
できた空間群が正解)。
データの転送
立体構造解析を実施するためには、反射データ(ScaleAveraged.ref)を、構造解析用PC“protein”へ転送する必要がある。
以下、その手順を示す。
なお、反射データと併せてdtscaleaverage.logを転送しておくと、結晶系の確認などに便利。
-
構造解析用PC“protein”を起動。
-
“protein2”ディスクトップ“Network”→“lebra - protein-structure (Protein)”をクリック。
-
ScaleAveraged.ref(+dtscaleaverage.log)をドラッグ&ドロップ。
⇒以上で、転送完了。
*“protein”の“LEBRA/protein-DataShare”フォルダ内にファイルが保存されている。