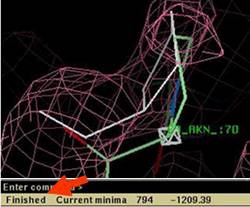
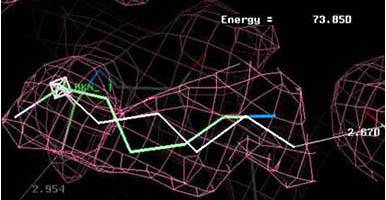
“Build atoms”パレット(図2.2.1)は、アミノ酸残基ごと、あるいは原子ごとに修正を行えるプログラムが表示されている。
ここでは各プログラムの説明の前に、パレット上で行う修正結果の保存方法について説明する。
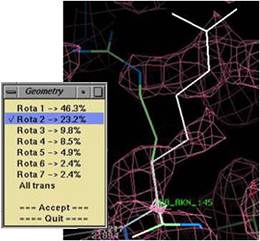
“Accept/Quit”パレット(図2.2.2)
後述の各修正プログラムを実行すると、修正結果が白・緑等の構造で表示され、併せて“Accept/Quit”パレットが表示される。
*“Fit Side chain by RSR”など、表示されないものもある。
この結果を受け入れるか否かを、同パレットで決定する。
Acceptした結果に問題がなければ、次の「Save changes」で保存。
取り消す場合は、「Undo last fit」
“Save changes”
“Build atoms”パレット“Save changes”(図2.2.1赤丸)をクリックすると、それまでにAcceptした結果が保存される。
こまめに「Save changes」をしたほうが無難。
“Undo last fit”
“Build atoms”パレットの“Undo last fit”(図2.2.1赤丸)をクリックすると、Acceptした結果が破棄される。
なお、既に「Save changes」済みの結果は破棄できない。
なお、“Save changes”は、X-Build実行中の結果を保存している状態。
最終的な修正内容の保存方法(=新規構造ファイルの作成)は「精密化の終了」に記述している。

(図2.2.1) “Build Atoms”パレット

(図2.2.2)“Accept/Quit”パレット